Jason Larkin's PhD Projects
Predicting Alloy Vibrational Mode Properties using Lattice Dynamics Calculations, Molecular Dynamics Simulations, and the Virtual Crystal Approximation
We investigated numerically the virtual crystal (VC) approximation for predicting the vibrational mode properties and thermal conductivity of alloys by examining a model for silicon/ germanium (SiGe) alloys. Due to their potentially low thermal conductivities, disordered materials (e.g., alloys, amorphous solids, aerogels) are used in applications ranging from thermoelectric energy conversion to thermally insulating barriers. Alloys are a subgroup of disordered materials where the atomic positions follow a lattice structure but the constituent species are spatially random.
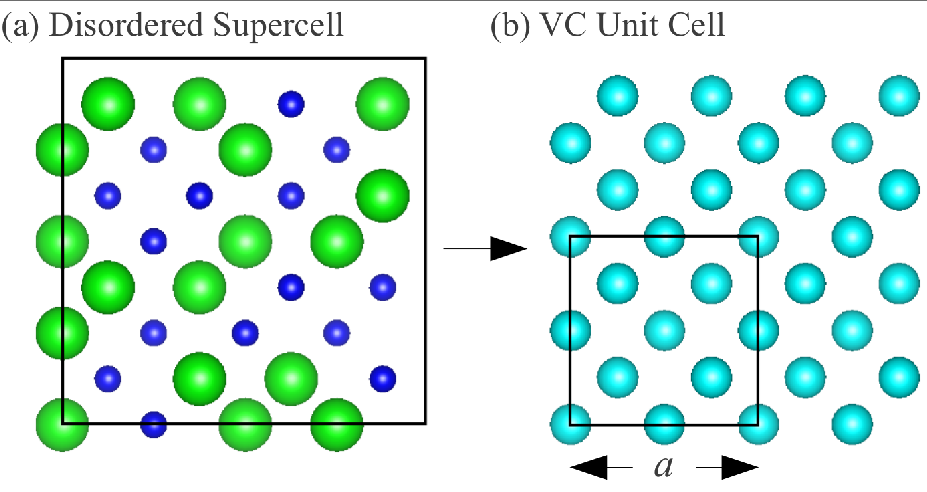
We study alloy disordered supercells with a range of concentrations from Si-rich (blue atoms):



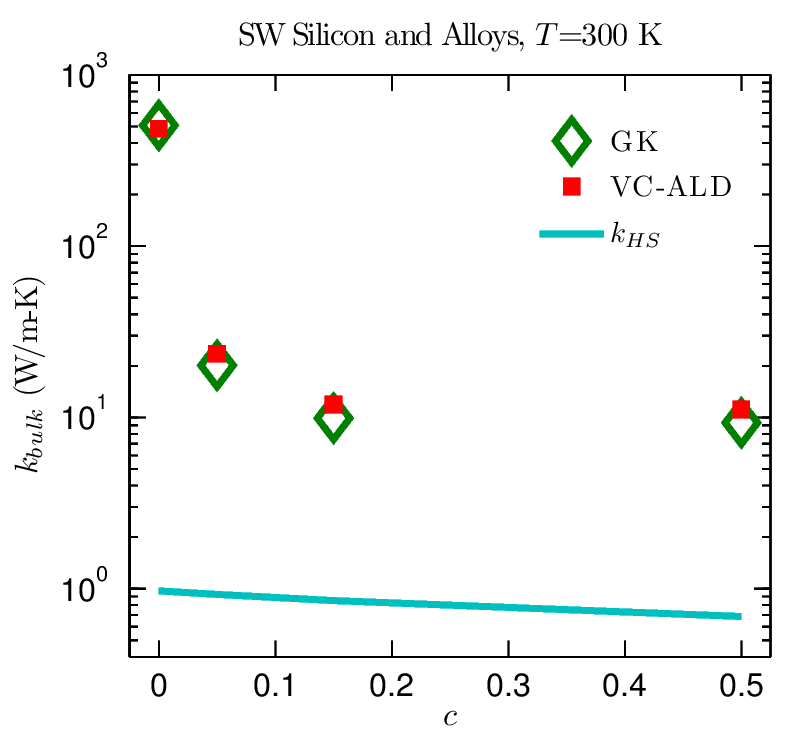
The above Figure shows thermal conductivity predictions for Si and SiGe alloys at a temperature of 300 K using the VC approximation (VC-ALD) and the top-down Green-Kubo method (GK). Good agreement between VC-ALD and GK is seen for SiGe, in part because the thermal conductivities for all concentrations are significantly higher than the high-scatter thermal conductivity prediction kHS. The results for the alloys studied suggest that modeling of thermal transport in ordered and disordered lattices can be separated into two broad groups: low- frequency dominated and full-spectrum materials.